publications
publications by categories in reversed chronological order.
2026
-
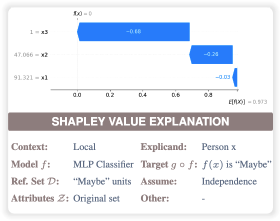 We need to explain our explanationsVenetia PliatsikaPreprint, 2026
We need to explain our explanationsVenetia PliatsikaPreprint, 2026Shapley values, derived from cooperative game theory, are widely used in machine learning to design, audit, and recommend recourse for complex models. A wide va- riety of implementations has resulted in explanation multiplicity, the phenomenon where multiple contradictory Shapley value explanations can be derived for the same model and data instance. In this position paper, we argue that an explanation is mathematically underspecified without a formal definition of context of use and a corresponding disclosure of technical assumptions. To enable both goals, we in- troduce a generalized definition of Shapley values and use it to reframe multiplicity as a parametrization problem. Using this unified parametrization, we propose a roadmap for the entire explanation lifecycle, calling focus on two primary research directions. First, we posit that context of use is a formal input to the explanatory process. As such, we require rigorous guidelines to map stakeholder questions to specific mathematical parameters. Second, we argue that an explanation is incomplete without a meta-explanation: a structured disclosure of the underlying assumptions and implementation choices used in its construction. As a step towards this second direction, we propose the Shapley Value Explainability Card. Finally, we demonstrate through several examples how each parameter choice implies specific mathematical assumptions about the data or the explanation’s intended use, and provide a detailed call to action towards a more transparent and context-aware explanation lifecycle
2025
-
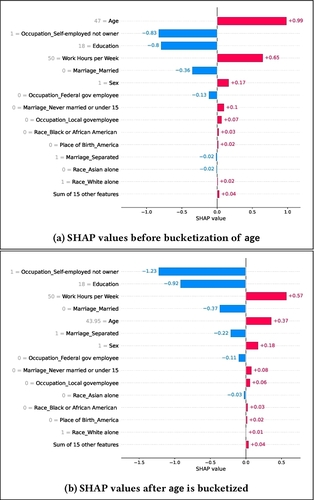 Shap-based explanations are sensitive to feature representationHyunseung Hwang, Andrew Bell, Joao Fonseca, and 3 more authorsIn Proceedings of the 2025 ACM Conference on Fairness, Accountability, and Transparency, 2025
Shap-based explanations are sensitive to feature representationHyunseung Hwang, Andrew Bell, Joao Fonseca, and 3 more authorsIn Proceedings of the 2025 ACM Conference on Fairness, Accountability, and Transparency, 2025Local feature-based explanations are a key component of the XAI toolkit. These explanations compute feature importance values relative to an “interpretable” feature representation. In tabular data, feature values themselves are often considered interpretable. This paper examines the impact of data engineering choices on local feature-based explanations. We demonstrate that simple, common data engineering techniques, such as representing age with a histogram or encoding race in a specific way, can manipulate feature importance as determined by popular methods like SHAP. Notably, the sensitivity of explanations to feature representation can be exploited by adversaries to obscure issues like discrimination. While the intuition behind these results is straightforward, their systematic exploration has been lacking. Previous work has focused on adversarial attacks on feature-based explainers by biasing data or manipulating models. To the best of our knowledge, this is the first study demonstrating that explainers can be misled by standard, seemingly innocuous data engineering techniques.
2024
-
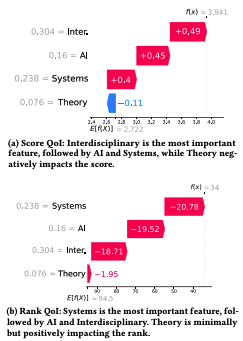 ShaRP: Explaining Rankings and Preferences with Shapley ValuesVenetia Pliatsika, Joao Fonseca, Kateryna Akhynko, and 2 more authorsarXiv preprint arXiv:2401.16744Published in VLDB 2025 , 2024
ShaRP: Explaining Rankings and Preferences with Shapley ValuesVenetia Pliatsika, Joao Fonseca, Kateryna Akhynko, and 2 more authorsarXiv preprint arXiv:2401.16744Published in VLDB 2025 , 2024Algorithmic decisions in critical domains such as hiring, college admissions, and lending are often based on rankings. Given the impact of these decisions on individuals, organizations, and population groups, it is essential to understand them - to help individuals improve their ranking position, design better ranking procedures, and ensure legal compliance. In this paper, we argue that explainability methods for classification and regression, such as SHAP, are insufficient for ranking tasks, and present ShaRP - Shapley Values for Rankings and Preferences - a framework that explains the contributions of features to various aspects of a ranked outcome. ShaRP computes feature contributions for various ranking-specific profit functions, such as rank and top-k, and also includes a novel Shapley value-based method for explaining pairwise preference outcomes. We provide a flexible implementation of ShaRP, capable of efficiently and comprehensively explaining ranked and pairwise outcomes over tabular data, in score-based ranking and learning-to-rank tasks. Finally, we develop a comprehensive evaluation methodology for ranking explainability methods, showing through qualitative, quantitative, and usability studies that our rank-aware QoIs offer complementary insights, scale effectively, and help users interpret ranked outcomes in practice.
-
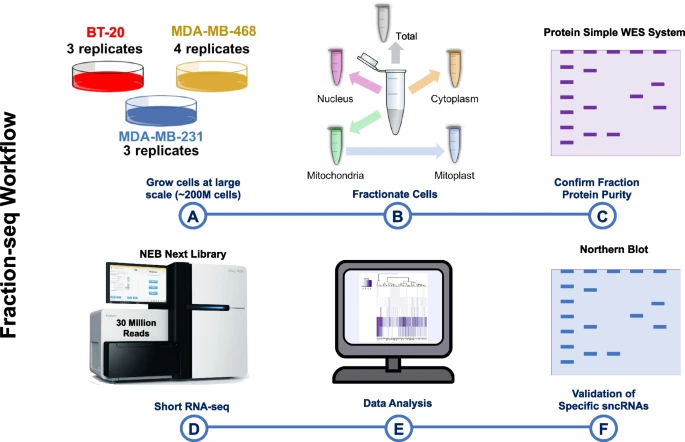 The subcellular distribution of miRNA isoforms, tRNA-derived fragments, and rRNA-derived fragments depends on nucleotide sequence and cell typeTess Cherlin, Yi Jing, Siddhartha Shah, and 8 more authorsBMC biology, 2024
The subcellular distribution of miRNA isoforms, tRNA-derived fragments, and rRNA-derived fragments depends on nucleotide sequence and cell typeTess Cherlin, Yi Jing, Siddhartha Shah, and 8 more authorsBMC biology, 2024Background: MicroRNA isoforms (isomiRs), tRNA-derived fragments (tRFs), and rRNA-derived fragments (rRFs) represent most of the small non-coding RNAs (sncRNAs) found in cells. Members of these three classes modulate messenger RNA (mRNA) and protein abundance and are dysregulated in diseases. Experimental studies to date have assumed that the subcellular distribution of these molecules is well-understood, independent of cell type, and the same for all isoforms of a sncRNA. Results: We tested these assumptions by investigating the subcellular distribution of isomiRs, tRFs, and rRFs in biological replicates from three cell lines from the same tissue and same-sex donors that model the same cancer subtype. In each cell line, we profiled the isomiRs, tRFs, and rRFs in the nucleus, cytoplasm, whole mitochondrion (MT), mitoplast (MP), and whole cell. Using a rigorous mathematical model we developed, we accounted for cross-fraction contamination and technical errors and adjusted the measured abundances accordingly. Analyses of the adjusted abundances show that isomiRs, tRFs, and rRFs exhibit complex patterns of subcellular distributions. These patterns depend on each sncRNA’s exact sequence and the cell type. Even in the same cell line, isoforms of the same sncRNA whose sequences differ by a few nucleotides (nts) can have different subcellular distributions. Conclusions: SncRNAs with similar sequences have different subcellular distributions within and across cell lines, suggesting that each isoform could have a different function. Future computational and experimental studies of isomiRs, tRFs, and rRFs will need to distinguish among each molecule’s various isoforms and account for differences in each isoform’s subcellular distribution in the cell line at hand. While the findings add to a growing body of evidence that isomiRs, tRFs, rRFs, tRNAs, and rRNAs follow complex intracellular trafficking rules, further investigation is needed to exclude alternative explanations for the observed subcellular distribution of sncRNAs.
-
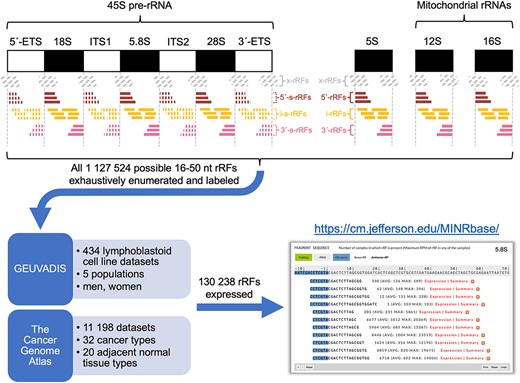 MINRbase: a comprehensive database of nuclear-and mitochondrial-ribosomal-RNA-derived fragments (rRFs)Venetia Pliatsika, Tess Cherlin, Phillipe Loher, and 4 more authorsNucleic Acids Research, 2024
MINRbase: a comprehensive database of nuclear-and mitochondrial-ribosomal-RNA-derived fragments (rRFs)Venetia Pliatsika, Tess Cherlin, Phillipe Loher, and 4 more authorsNucleic Acids Research, 2024We describe the Mitochondrial and Nuclear rRNA fragment database (MINRbase), a knowledge repository aimed at facilitating the study of ribosomal RNA-derived fragments (rRFs). MINRbase provides interactive access to the profiles of 130 238 expressed rRFs arising from the four human nuclear rRNAs (18S, 5.8S, 28S, 5S), two mitochondrial rRNAs (12S, 16S) or four spacers of 45S pre-rRNA. We compiled these profiles by analyzing 11 632 datasets, including the GEUVADIS and The Cancer Genome Atlas (TCGA) repositories. MINRbase offers a user-friendly interface that lets researchers issue complex queries based on one or more criteria, such as parental rRNA identity, nucleotide sequence, rRF minimum abundance and metadata keywords (e.g. tissue type, disease). A ‘summary’ page for each rRF provides a granular breakdown of its expression by tissue type, disease, sex, ancestry and other variables; it also allows users to create publication-ready plots at the click of a button. MINRbase has already allowed us to generate support for three novel observations: the internal spacers of 45S are prolific producers of abundant rRFs; many abundant rRFs straddle the known boundaries of rRNAs; rRF production is regimented and depends on ‘personal attributes’ (sex, ancestry) and ‘context’ (tissue type, tissue state, disease). MINRbase is available at https://cm.jefferson.edu/MINRbase/.
2021
-
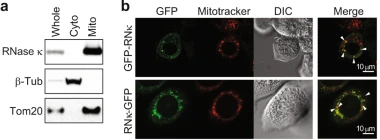 RNase κpromotes robust piRNA production by generating 2′, 3′-cyclic phosphate-containing precursorsMegumi Shigematsu, Takuya Kawamura, Keisuke Morichika, and 8 more authorsNature Communications, 2021
RNase κpromotes robust piRNA production by generating 2′, 3′-cyclic phosphate-containing precursorsMegumi Shigematsu, Takuya Kawamura, Keisuke Morichika, and 8 more authorsNature Communications, 2021In animal germlines, PIWI proteins and the associated PIWI-interacting RNAs (piRNAs) protect genome integrity by silencing transposons. Here we report the extensive sequence and quantitative correlations between 2′,3′-cyclic phosphate-containing RNAs (cP-RNAs), identified using cP-RNA-seq, and piRNAs in the Bombyx germ cell line and mouse testes. The cP-RNAs containing 5′-phosphate (P-cP-RNAs) identified by P-cP-RNA-seq harbor highly consistent 5′-end positions as the piRNAs and are loaded onto PIWI protein, suggesting their direct utilization as piRNA precursors. We identified Bombyx RNase Kappa (BmRNase κ) as a mitochondria-associated endoribonuclease which produces cP-RNAs during piRNA biogenesis. BmRNase κ-depletion elevated transposon levels and disrupted a piRNA-mediated sex determination in Bombyx embryos, indicating the crucial roles of BmRNase κ in piRNA biogenesis and embryonic development. Our results reveal a BmRNase κ-engaged piRNA biogenesis pathway, in which the generation of cP-RNAs promotes robust piRNA production.
-
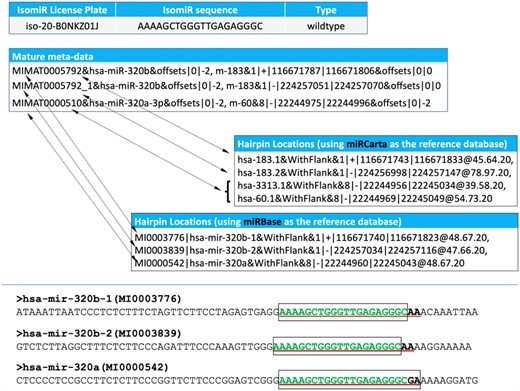 IsoMiRmap: fast, deterministic and exhaustive mining of isomiRs from short RNA-seq datasetsPhillipe Loher, Nestoras Karathanasis, Eric Londin, and 4 more authorsBioinformatics, 2021
IsoMiRmap: fast, deterministic and exhaustive mining of isomiRs from short RNA-seq datasetsPhillipe Loher, Nestoras Karathanasis, Eric Londin, and 4 more authorsBioinformatics, 2021Motivation: MicroRNA (miRNA) precursor arms give rise to multiple isoforms simultaneously called ‘isomiRs.’ IsomiRs from the same arm typically differ by a few nucleotides at either their 5′ or 3′ termini or both. In humans, the identities and abundances of isomiRs depend on a person’s sex and genetic ancestry as well as on tissue type, tissue state and disease type/subtype. Moreover, nearly half of the time the most abundant isomiR differs from the miRNA sequence found in public databases. Accurate mining of isomiRs from deep sequencing data is thus important. Results: We developed isoMiRmap, a fast, standalone, user-friendly mining tool that identifies and quantifies all isomiRs by directly processing short RNA-seq datasets. IsoMiRmap is a portable ‘plug-and-play’ tool, requires minimal setup, has modest computing and storage requirements, and can process an RNA-seq dataset with 50 million reads in just a few minutes on an average laptop. IsoMiRmap deterministically and exhaustively reports all isomiRs in a given deep sequencing dataset and quantifies them accurately (no double-counting). IsoMiRmap comprehensively reports all miRNA precursor locations from which an isomiR may be transcribed, tags as ‘ambiguous’ isomiRs whose sequences exist both inside and outside of the space of known miRNA sequences and reports the public identifiers of common single-nucleotide polymorphisms and documented somatic mutations that may be present in an isomiR. IsoMiRmap also identifies isomiRs with 3’ non-templated post-transcriptional additions. Compared to similar tools, isoMiRmap is the fastest, reports more bona fide isomiRs, and provides the most comprehensive information related to an isomiR’s transcriptional origin. Availability and implementation: The codes for isoMiRmap are freely available at https://cm.jefferson.edu/isoMiRmap/ and https://github.com/TJU-CMC-Org/isoMiRmap/. IsomiR profiles for the datasets of the 1000 Genomes Project, spanning five population groups, and The Cancer Genome Atlas (TCGA), spanning 33 cancer studies, are also available at https://cm.jefferson.edu/isoMiRmap/. Supplementary information: Supplementary data are available at Bioinformatics online.
2020
-
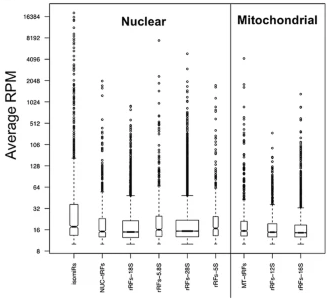 Ribosomal RNA fragmentation into short RNAs (rRFs) is modulated in a sex-and population of origin-specific mannerTess Cherlin, Rogan Magee, Yi Jing, and 3 more authorsBmc Biology, 2020
Ribosomal RNA fragmentation into short RNAs (rRFs) is modulated in a sex-and population of origin-specific mannerTess Cherlin, Rogan Magee, Yi Jing, and 3 more authorsBmc Biology, 2020Background: The advent of next generation sequencing (NGS) has allowed the discovery of short and long non-coding RNAs (ncRNAs) in an unbiased manner using reverse genetics approaches, enabling the discovery of multiple categories of ncRNAs and characterization of the way their expression is regulated. We previously showed that the identities and abundances of microRNA isoforms (isomiRs) and transfer RNA-derived fragments (tRFs) are tightly regulated, and that they depend on a person’s sex and population origin, as well as on tissue type, tissue state, and disease type. Here, we characterize the regulation and distribution of fragments derived from ribosomal RNAs (rRNAs). rRNAs form a group that includes four (5S, 5.8S, 18S, 28S) rRNAs encoded by the human nuclear genome and two (12S, 16S) by the mitochondrial genome. rRNAs constitute the most abundant RNA type in eukaryotic cells. Results: We analyzed rRNA-derived fragments (rRFs) across 434 transcriptomic datasets obtained from lymphoblastoid cell lines (LCLs) derived from healthy participants of the 1000 Genomes Project. The 434 datasets represent five human populations and both sexes. We examined each of the six rRNAs and their respective rRFs, and did so separately for each population and sex. Our analysis shows that all six rRNAs produce rRFs with unique identities, normalized abundances, and lengths. The rRFs arise from the 5′-end (5′-rRFs), the interior (i-rRFs), and the 3′-end (3′-rRFs) or straddle the 5′ or 3′ terminus of the parental rRNA (x-rRFs). Notably, a large number of rRFs are produced in a population-specific or sex-specific manner. Preliminary evidence suggests that rRF production is also tissue-dependent. Of note, we find that rRF production is not affected by the identity of the processing laboratory or the library preparation kit. Conclusions: Our findings suggest that rRFs are produced in a regimented manner by currently unknown processes that are influenced by both ubiquitous as well as population-specific and sex-specific factors. The properties of rRFs mirror the previously reported properties of isomiRs and tRFs and have implications for the study of homeostasis and disease.
2019
-
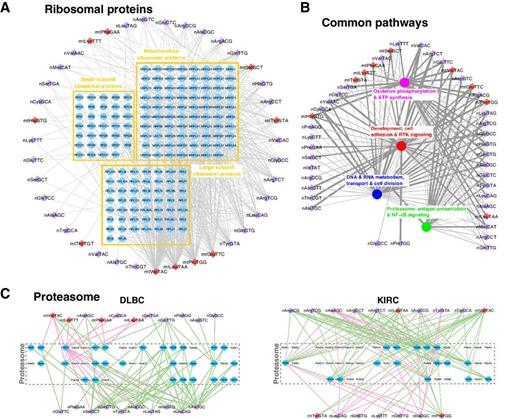 tRNA fragments show intertwining with mRNAs of specific repeat content and have links to disparitiesAristeidis G Telonis, Phillipe Loher, Rogan Magee, and 4 more authorsCancer research, 2019
tRNA fragments show intertwining with mRNAs of specific repeat content and have links to disparitiesAristeidis G Telonis, Phillipe Loher, Rogan Magee, and 4 more authorsCancer research, 2019tRNA-derived fragments (tRF) are a class of potent regulatory RNAs. We mined the datasets from The Cancer Genome Atlas (TCGA) representing 32 cancer types with a deterministic and exhaustive pipeline for tRNA fragments. We found that mitochondrial tRNAs contribute disproportionally more tRFs than nuclear tRNAs. Through integrative analyses, we uncovered a multitude of statistically significant and context-dependent associations between the identified tRFs and mRNAs. In many of the 32 cancer types, these associations involve mRNAs from developmental processes, receptor tyrosine kinase signaling, the proteasome, and metabolic pathways that include glycolysis, oxidative phosphorylation, and ATP synthesis. Even though the pathways are common to multiple cancers, the association of specific mRNAs with tRFs depends on and differs from cancer to cancer. The associations between tRFs and mRNAs extend to genomic properties as well; specifically, tRFs are positively correlated with shorter genes that have a higher density in repeats, such as ALUs, MIRs, and ERVLs. Conversely, tRFs are negatively correlated with longer genes that have a lower repeat density, suggesting a possible dichotomy between cell proliferation and differentiation. Analyses of bladder, lung, and kidney cancer data indicate that the tRF-mRNA wiring can also depend on a patient’s sex. Sex-dependent associations involve cyclin-dependent kinases in bladder cancer, the MAPK signaling pathway in lung cancer, and purine metabolism in kidney cancer. Taken together, these findings suggest diverse and wide-ranging roles for tRFs and highlight the extensive interconnections of tRFs with key cellular processes and human genomic architecture. Significance: Across 32 TCGA cancer contexts, nuclear and mitochondrial tRNA fragments exhibit associations with mRNAs that belong to concrete pathways, encode proteins with particular destinations, have a biased repeat content, and are sex dependent.
2018
-
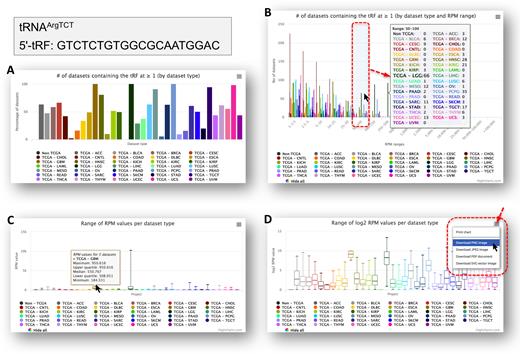 MINTbase v2. 0: a comprehensive database for tRNA-derived fragments that includes nuclear and mitochondrial fragments from all The Cancer Genome Atlas projectsVenetia Pliatsika, Phillipe Loher, Rogan Magee, and 5 more authorsNucleic acids research, 2018
MINTbase v2. 0: a comprehensive database for tRNA-derived fragments that includes nuclear and mitochondrial fragments from all The Cancer Genome Atlas projectsVenetia Pliatsika, Phillipe Loher, Rogan Magee, and 5 more authorsNucleic acids research, 2018MINTbase is a repository that comprises nuclear and mitochondrial tRNA-derived fragments (‘tRFs’) found in multiple human tissues. The original version of MINTbase comprised tRFs obtained from 768 transcriptomic datasets. We used our deterministic and exhaustive tRF mining pipeline to process all of The Cancer Genome Atlas datasets (TCGA). We identified 23 413 tRFs with abundance of ≥ 1.0 reads-per-million (RPM). To facilitate further studies of tRFs by the community, we just released version 2.0 of MINTbase that contains information about 26 531 distinct human tRFs from 11 719 human datasets as of October 2017. Key new elements include: the ability to filter tRFs on-the-fly by minimum abundance thresholding; the ability to filter tRFs by tissue keywords; easy access to information about a tRF’s maximum abundance and the datasets that contain it; the ability to generate relative abundance plots for tRFs across cancer types and convert them into embeddable figures; MODOMICS information about modifications of the parental tRNA, etc. Version 2.0 of MINTbase contains 15x more datasets and nearly 4x more distinct tRFs than the original version, yet continues to offer fast, interactive access to its contents. Version 2.0 is available freely at http://cm.jefferson.edu/MINTbase/.
2017
-
 Profit sharing and efficiency in utility gamesSreenivas Gollapudi, Kostas Kollias, Debmalya Panigrahi, and 1 more authorIn 25th Annual European Symposium on Algorithms (ESA 2017), 2017
Profit sharing and efficiency in utility gamesSreenivas Gollapudi, Kostas Kollias, Debmalya Panigrahi, and 1 more authorIn 25th Annual European Symposium on Algorithms (ESA 2017), 2017We study utility games (Vetta, FOCS 2002) where a set of players join teams to produce social utility, and receive individual utility in the form of payments in return. These games have many natural applications in competitive settings such as labor markets, crowdsourcing, etc. The efficiency of such a game depends on the profit sharing mechanism - the rule that maps utility produced by the players to their individual payments. We study three natural and widely used profit sharing mechanisms - egalitarian or equal sharing, marginal gain or value addition when a player joins, and marginal loss or value depletion when a player leaves. For these settings, we give tight bounds on the price of anarchy, thereby allowing comparison between these popular mechanisms from a (worst case) social welfare perspective.
2016
-
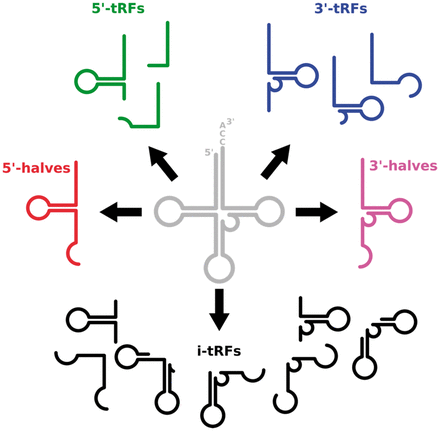 MINTbase: a framework for the interactive exploration of mitochondrial and nuclear tRNA fragmentsVenetia Pliatsika, Phillipe Loher, Aristeidis G Telonis, and 1 more authorBioinformatics, 2016
MINTbase: a framework for the interactive exploration of mitochondrial and nuclear tRNA fragmentsVenetia Pliatsika, Phillipe Loher, Aristeidis G Telonis, and 1 more authorBioinformatics, 2016Motivation: It has been known that mature transfer RNAs (tRNAs) that are encoded in the nuclear genome give rise to short molecules, collectively known as tRNA fragments or tRFs. Recently, we reported that, in healthy individuals and in patients, tRFs are constitutive, arise from mitochondrial as well as from nuclear tRNAs, and have composition and abundances that depend on a person’s sex, population origin and race as well as on tissue, disease and disease subtype. Our findings as well as similar work by other groups highlight the importance of tRFs and presage an increase in the community’s interest in elucidating the roles of tRFs in health and disease. Results: We created MINTbase, a web-based framework that serves the dual-purpose of being a content repository for tRFs and a tool for the interactive exploration of these newly discovered molecules. A key feature of MINTbase is that it deterministically and exhaustively enumerates all possible genomic locations where a sequence fragment can be found and indicates which fragments are exclusive to tRNA space, and thus can be considered as tRFs: this is a very important consideration given that the genomes of higher organisms are riddled with partial tRNA sequences and with tRNA-lookalikes whose aberrant transcripts can be mistaken for tRFs. MINTbase is extremely flexible and integrates and presents tRF information from multiple yet interconnected vantage points (‘vistas’). Vistas permit the user to interactively personalize the information that is returned and the manner in which it is displayed. MINTbase can report comparative information on how a tRF is distributed across all anticodon/amino acid combinations, provides alignments between a tRNA and multiple tRFs with which the user can interact, provides details on published studies that reported a tRF as expressed, etc. Importantly, we designed MINTbase to contain all possible tRFs that could ever be produced by mature tRNAs: this allows us to report on their genomic distributions, anticodon/amino acid properties, alignments, etc. while giving users the ability to at-will investigate candidate tRF molecules before embarking on focused experimental explorations. Lastly, we also introduce a new labeling scheme that is tRF-sequence-based and allows users to associate a tRF with a universally unique label (‘tRF-license plate’) that is independent of a genome assembly and does not require any brokering mechanism.
2015
-
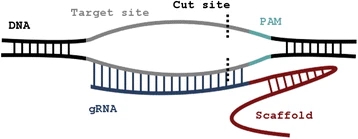 “Off-Spotter”: very fast and exhaustive enumeration of genomic lookalikes for designing CRISPR/Cas guide RNAsVenetia Pliatsika and Isidore RigoutsosBiology direct, 2015
“Off-Spotter”: very fast and exhaustive enumeration of genomic lookalikes for designing CRISPR/Cas guide RNAsVenetia Pliatsika and Isidore RigoutsosBiology direct, 2015Background: CRISPR/Cas (Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR associated nucleases) is a powerful component of the prokaryotic immune system that has been adapted for targeted genetic engineering in higher organisms. A key element of CRISPR/Cas is the “guide” RNA (gRNA) that is 20 nucleotides (nts) in length and designed to be complementary to the intended target site. An integral requirement of the CRISPR/Cas system is that the target site be followed by a protospacer adjacent motif (PAM). Care needs to be exercised during gRNA design to avoid unintended (“off-target”) interactions. Results: We designed and implemented the Off-Spotter algorithm to assist with the design of optimal gRNAs. When presented with a candidate gRNA sequence and a PAM, Off-Spotter quickly and exhaustively identifies all genomic sites that satisfy the PAM constraint and are identical or nearly-identical to the provided gRNA. Off-Spotter achieves its extreme performance through purely algorithmic means and not through hardware accelerators such as graphical processing units (GPUs). Off-Spotter also allows the user to identify on-the-fly how many and which nucleotides of the gRNA comprise the “seed”. Off-Spotter’s output includes a histogram showing the number of potential off-targets as a function of the number of mismatches. The output also includes for each potential off-target the site’s genomic location, a human genome browser hyperlink to the corresponding location, genomic annotation in the vicinity of the off-target, GC content, etc. Conclusion: Off-Spotter is very fast and flexible and can help in the design of optimal gRNAs by providing several PAM choices, a run-time definition of the seed and of the allowed number of mismatches, and a flexible output interface that allows sorting of the results, optional viewing/hiding of columns, etc. A key element of Off-Spotter is that it does not have a rigid definition of the seed: instead, the user can declare both the seed’s location and extent on-the-fly. We expect that this flexibility in combination with Off-Spotter’s speed and richly annotated output will enable experimenters to interactively and quickly explore different scenarios and gRNA possibilities. Reviewed: This article was reviewed by Dr Eugene Koonin and Dr Frank Eisenhaber.